|
Cracking the regulatory genome: Which genetic variants in DNase I sensitive regions are functional? |
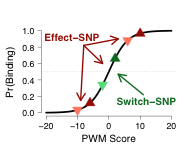
We developed a novel approach to annotating functional regulatory regions that integrates sequence information (e.g., position weight matrices) with DNaseI footprinting data to predict the impact of a sequence change on TF binding. We identified over 3 million regulatory variants predicted to affect active regulatory regions for a panel of TFs in over 650 cell-types.
Moyerbrailean GA, Kalita CA, Harvey CT, Wen X, Luca F, Pique-Regi R. (2016) Which Genetics Variants in DNase-Seq Footprints Are More Likely to Alter Binding? PLoS Genet 12(2): e1005875. doi: 10.1371/journal.pgen.1005875
We have provided annotations as a brower hub to facilitate viewing of the data. There are two tracks: 1) regulatory variants, and 2) regulatory footprints.
CentiSNPs Compendium
For each transcription factor motif we summarized all SNPs in footprints accross all ENCODE and Roadmap Epigenome (see README in the link below for more details)
Regulatory Regions
The footprint annotation data is also available as bed files, separated both by cell-type and TF motif. Each file is formatted as bed9+ files, with four additional data fields: SNP position, 1KG reference allele, 1KG alternate allele, prior log ratio for binding of the reference allele, and the prior LR for binding of the alternate allele. For TFBS without a polymorphism, the SNP position is listed as "0" and the prior LR for the reference allele is repeated.
Supplemental Files
The supplemental excel files associated with the above publications can be downloaded here:
Additional Files
For questions related to the hub or the data, please contact: